進行性脊肌萎縮癥
進行性脊肌萎縮癥百科
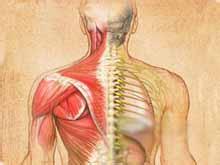
進行性脊肌萎縮癥發病年齡稍早與ALS,多在30歲左右,男性多見,運動神經元變性僅限於脊髓前角細胞,表現肌無力、肌萎縮和肌束震顫等下運動神經元受損癥狀體征,隱襲起病,首發癥狀常為一手或雙手小肌肉萎縮、無力,逐漸累及前臂、上臂及肩胛帶肌肉,也有從下肢萎縮開始者,但少見,遠端萎縮明顯,肌張力及腱反射減低,無感覺障礙,括約肌功能不受累,累及延髓出現延髓麻痹者存活時間段,常死於肺感染.

進行性脊肌萎縮癥
進行性脊肌萎縮癥病因
進行性脊肌萎縮僅由脊髓前角細胞變性所致.雖經許多研究,提出過慢病毒感染、免疫功能異常、遺傳因素、重金屬中毒、營養代謝障礙以及環境等因素致病的假說,但均未被證實.

進行性脊肌萎縮癥
進行性脊肌萎縮癥症状
多在30歲,男性多見,運動神經元變性僅限於脊髓前角細胞,表現肌無力、肌萎縮和肌束震顫等下運動神經元受損體征,隱襲起病,首發常為一手或雙手小肌肉萎縮、無力,逐漸累及前臂、上臂及肩胛帶肌肉,也有從下肢萎縮者,但少見,遠端萎縮,肌張力及腱反射減低,無障礙,括約肌功能不受累,累及延髓出現延髓麻痹者存活段,常死於肺感染.
進行性脊肌萎縮癥
進行性脊肌萎縮癥检查
進行性脊肌萎縮癥的檢查化驗
1、神經電生理:肌電圖呈典型神經源性改變.靜息狀態下可見纖顫電位、正銳播,有時可見束顫電位;小力收縮時運動單位電位時限增寬、波幅增大、多相波增加,大力收縮呈現單純相.神經傳導.運動誘發電位有助於確定上運動神經元損害.
2、肌肉活檢:有助於,但無特異性,早期為神經源性肌萎縮,晚期在光鏡下與肌源性肌萎縮不易鑒別.
3、血生化、CSF多無異常,肌酸磷酸激酶(CK)活性可輕度異常,MRI可顯示部分病例受累脊髓和腦幹萎縮變小.
進行性脊肌萎縮癥的鑒別診斷
大多數病例都屬常染色體隱性遺傳,看來都是第5號染色體上一個單獨的基因位點上的等位基因突變.有四種主要的變型.
Ⅰ型脊肌萎縮癥(Werdnig-Hoffmann病)在胎兒中已存在或在出生後2~4個月出現癥狀.大多數患病嬰兒在出生時就有肌張力過低的表現;在6個月齡期前,所有患病嬰兒都已表現出明顯的運動功能發育的延緩.95%的病孩在1歲前後死亡,沒有病例能存活超過4歲的,通常都是死於呼吸衰竭.
Ⅱ型(中間型)脊肌萎縮癥患兒大多數是在6~12個月期間出現癥狀,在2歲以前所有病例都已有明顯癥狀.不到25%的病例能學會坐,但沒有能走或能爬的.所有患兒都顯出肌張力過低,伴松弛性肌肉無力,腱反射消失與肌肉束顫,後者在幼兒中不容易察覺.可有吞咽困難.患兒往往因呼吸道並發癥在早年夭折,但也有病情進展自發停頓的,使患兒處於永久性非進展性的無力狀態中.
Ⅲ型脊肌萎縮癥(Wohlfart-Kugelberg-Welander病)在2~30歲期間發病.病理變化及遺傳方式與前兩種變型相似,但病情進展較為緩慢,預期壽命也較長.腿部的無力與肌萎縮最為顯著,以股四頭肌與髖關節屈肌最早出現癥狀.較後可累及臂部.無力現象往往從近端向遠端擴展.某些傢族性病例可能是繼發於特殊的酶的缺陷(例如氨基己糖苷酯酶缺乏).
Ⅳ型脊肌萎縮癥遺傳方式不定(常染色體隱性,常染色體顯性,性聯),成年期發病(年齡30~60歲),病情進展緩慢.可能無法將其與肌萎縮性側索硬化癥的下體育運動是人們遵循人體的生長發育規律和身體的活動規律,通過身體鍛煉、技術、訓練、競技比賽等方式達到增強體質,提高運動技術水平,豐富文化生活為目的的社會活動.
進行性脊肌萎縮癥预防
SMA產前診斷是隨著SMA基因研究的深入而開展.該法優點是在沒有取得先證者標本的傢系中也可以進行產前診斷.必要時應終止妊娠.





























